InVivoMAb anti-mouse TNFR2 (CD120b)
Product Details
The TR75-54.7 monoclonal antibody reacts with mouse Tumor Necrosis Factor Receptor Type II (TNFR2) also known as CD120b, TNFR type II, and p75. TNFR2 is expressed on many cell types at low levels; upon activation the expression is upregulated. Upon binding either of its two ligands, TNFα or LTα (lymphotoxin alpha) TNFR2 signal transduction leads to a wide spectrum of biological processes including immunoregulation, cell proliferation, differentiation, apoptosis, NF-kB activation, increased expression of proinflammatory genes, antitumor activity, inflammation, anorexia, cachexia, septic shock, hematopoiesis, and viral replication. The TR75-54.7 antibody has been reported to block ligand-induced receptor signaling.Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Recombinant mouse TNFR2 |
| Reported Applications |
in vivo TNFR2 blockade in vitro TNFR2 blockade |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_2687728 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb polyclonal Armenian hamster IgG
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo TNFR2 blockade
Mansouri, S., et al. (2020). "Lung IFNAR1(hi) TNFR2(+) cDC2 promotes lung regulatory T cells induction and maintains lung mucosal tolerance at steady state" Mucosal Immunol 13(4): 595-608. PubMed
The lung is a naturally tolerogenic organ. Lung regulatory T cells (T-regs) control lung mucosal tolerance. Here, we identified a lung IFNAR1(hi)TNFR2(+) conventional DC2 (iR2D2) population that induces T-regs in the lung at steady state. Using conditional knockout mice, adoptive cell transfer, receptor blocking antibodies, and TNFR2 agonist, we showed that iR2D2 is a lung microenvironment-adapted dendritic cell population whose residence depends on the constitutive TNFR2 signaling. IFNβ-IFNAR1 signaling in iR2D2 is necessary and sufficient for T-regs induction in the lung. The Epcam(+)CD45(-) epithelial cells are the sole lung IFNβ producer at the steady state. Surprisingly, iR2D2 is plastic. In a house dust mite model of asthma, iR2D2 generates lung T(H)2 responses. Last, healthy human lungs have a phenotypically similar tolerogenic iR2D2 population, which became pathogenic in lung disease patients. Our findings elucidate lung epithelial cells IFNβ-iR2D2-T-regs axis in controlling lung mucosal tolerance and provide new strategies for therapeutic interventions.
in vivo TNFR2 blockade
Hurrell, B. P., et al. (2019). "TNFR2 Signaling Enhances ILC2 Survival, Function, and Induction of Airway Hyperreactivity" Cell Rep 29(13): 4509-4524.e4505. PubMed
Group 2 innate lymphoid cells (ILC2s) can initiate pathologic inflammation in allergic asthma by secreting copious amounts of type 2 cytokines, promoting lung eosinophilia and airway hyperreactivity (AHR), a cardinal feature of asthma. We discovered that the TNF/TNFR2 axis is a central immune checkpoint in murine and human ILC2s. ILC2s selectively express TNFR2, and blocking the TNF/TNFR2 axis inhibits survival and cytokine production and reduces ILC2-dependent AHR. The mechanism of action of TNFR2 in ILC2s is through the non-canonical NF-κB pathway as an NF-κB-inducing kinase (NIK) inhibitor blocks the costimulatory effect of TNF-α. Similarly, human ILC2s selectively express TNFR2, and using hILC2s, we show that TNFR2 engagement promotes AHR through a NIK-dependent pathway in alymphoid murine recipients. These findings highlight the role of the TNF/TNFR2 axis in pulmonary ILC2s, suggesting that targeting TNFR2 or relevant signaling is a different strategy for treating patients with ILC2-dependent asthma.
in vivo TNFR2 blockade
Leclerc, M., et al. (2016). "Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells" Blood 128(12): 1651-1659. PubMed
Therapeutic CD4(+)Foxp3(+) natural regulatory T cells (Tregs) can control experimental graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation (allo-HCT) by suppressing conventional T cells (Tconvs). Treg-based therapies are currently tested in clinical trials with promising preliminary results in allo-HCT. Here, we hypothesized that as Tregs are capable of modulating Tconv response, it is likely that the inflammatory environment and particularly donor T cells are also capable of influencing Treg function. Indeed, previous findings in autoimmune diabetes revealed a feedback mechanism that renders Tconvs able to stimulate Tregs by a mechanism that was partially dependent on tumor necrosis factor (TNF). We tested this phenomenon during alloimmune response in our previously described model of GVHD protection using antigen specific Tregs. Using different experimental approaches, we observed that control of GVHD by Tregs was fully abolished by blocking TNF receptor type 2 (TNFR2) or by using TNF-deficient donor T cells or TNFR2-deficient Tregs. Thus, our results show that Tconvs exert a powerful modulatory activity on therapeutic Tregs and clearly demonstrate that the sole defect of TNF production by donor T cells was sufficient to completely abolish the Treg suppressive effect in GVHD. Importantly, our findings expand the understanding of one of the central components of Treg action, the inflammatory context, and support that targeting TNF/TNFR2 interaction represents an opportunity to efficiently modulate alloreactivity in allo-HCT to either exacerbate it for a powerful antileukemic effect or reduce it to control GVHD.
in vivo TNFR2 blockade, in vitro TNFR2 blockade
DeBerge, M. P., et al. (2015). "Shedding of TNF receptor 2 by effector CD8+ T cells by ADAM17 is important for regulating TNF-alpha availability during influenza infection" J Leukoc Biol 98(3): 423-434. PubMed
Elevated levels of solTNFR2 are observed in a variety of human pathophysiological conditions but regulation of TNFR2 levels during disease is not well understood. We found that solTNFR2 levels were increased following influenza infection or live-attenuated influenza virus challenge in mice and humans, respectively. As influenza-specific CD8(+) T cells up-regulated expression of TNFR2 after infection in mice, we hypothesized that CD8(+) T cells contributed, in part, to solTNFR2 production after influenza infection and were interested in the mechanisms by which CD8(+) T cells regulate TNFR2 shedding. Activation of these cells by TCR stimulation resulted in enhanced shedding of TNFR2 that required actin remodeling and lipid raft formation and was dependent on MAPK/ERK signaling. Furthermore, we identified ADAM17 as the protease responsible for TNFR2 shedding by CD8(+) T cells, with ADAM17 and TNFR2 required in “cis” for shedding to occur. We observed similar activation thresholds for TNF-alpha expression and TNFR2 shedding, suggesting that solTNFR2 functioned, in part, to regulate solTNF-alpha levels. Production of solTNFR2 by activated CD8(+) T cells reduced the availability of solTNF-alpha released by these cells, and TNFR2 blockade during influenza infection in mice enhanced the levels of solTNF-alpha, supporting this hypothesis. Taken together, this study identifies critical cellular mechanisms regulating TNFR2 shedding on CD8(+) T cells and demonstrates that TNFR2 contributes, in part, to the regulation of TNF-alpha levels during infection.
in vivo TNFR2 blockade
Bruggeman, L. A., et al. (2011). "TNFR2 interposes the proliferative and NF-kappaB-mediated inflammatory response by podocytes to TNF-alpha" Lab Invest 91(3): 413-425. PubMed
The development of proliferative podocytopathies has been linked to ligation of tumor necrosis factor receptor 2 (TNFR2) expressed on the renal parenchyma; however, the TNFR2-positive cells within the kidney responsible for podocyte injury are unknown. We detected de novo expression of TNFR2 on podocytes before hyperplastic injury in crescentic glomerulonephritis of mice with nephrotoxic nephritis, and in collapsing glomerulopathy of Tg26(HIV/nl) mice, kd/kd mice, and human beings. We further found that serum levels of soluble TNF-alpha and TNFR2 correlated significantly with renal injury in Tg26(HIV/nl) mice. Thus, we asked whether ligand binding of TNFR2 on podocytes ex vivo precipitates the characteristic proliferative and pro-inflammatory diseased podocyte phenotypes. Soluble TNF-alpha activated NF-kappaB and dose-dependently induced podocyte proliferation, marked by the expression of the podocyte G(1) cyclin and NF-kappaB target gene, cyclin D1. Microarray gene and chemokine protein expression profiling showed a marked pro-inflammatory NF-kappaB signature, and activated podocytes secreting CCL2- and CCL5-induced macrophage migration in transwell assays. Neutralization of TNFR2 on podocytes with blocking antibodies abrogated NF-kappaB activation and the induction of cyclin D1 by TNF-alpha, and identified TNFR2 as the primary receptor that induced IkappaBalpha degradation, the initiating event in NF-kappaB activation. These results suggest that TNFR2 expressed on podocytes and its canonical NF-kappaB signaling may directly interpose the compound pathogenic responses by podocytes to TNF-alpha, in the absence of other TNFR2-positive renal cell types in proliferative podocytopathies.
- In Vivo,
- Mus musculus (House mouse)
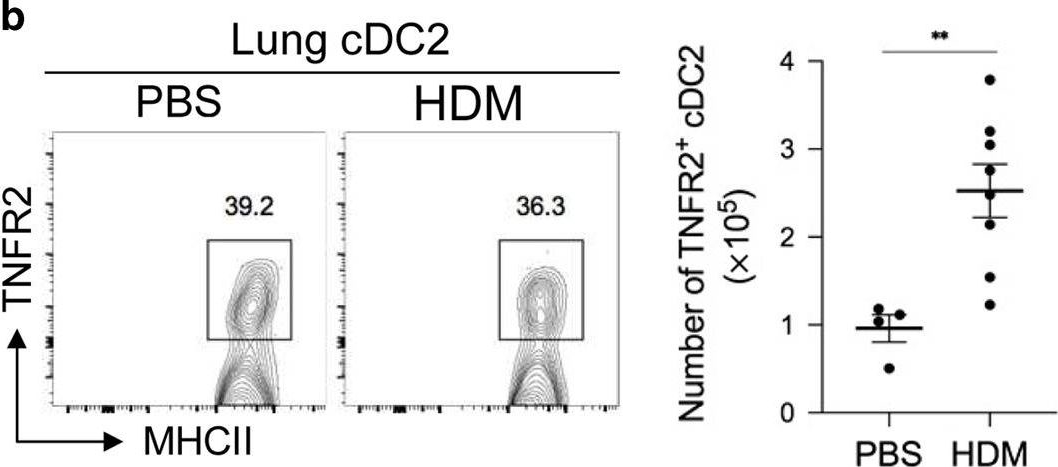
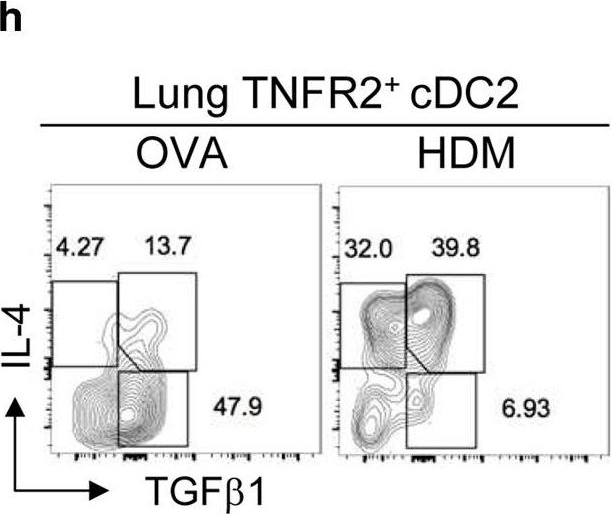
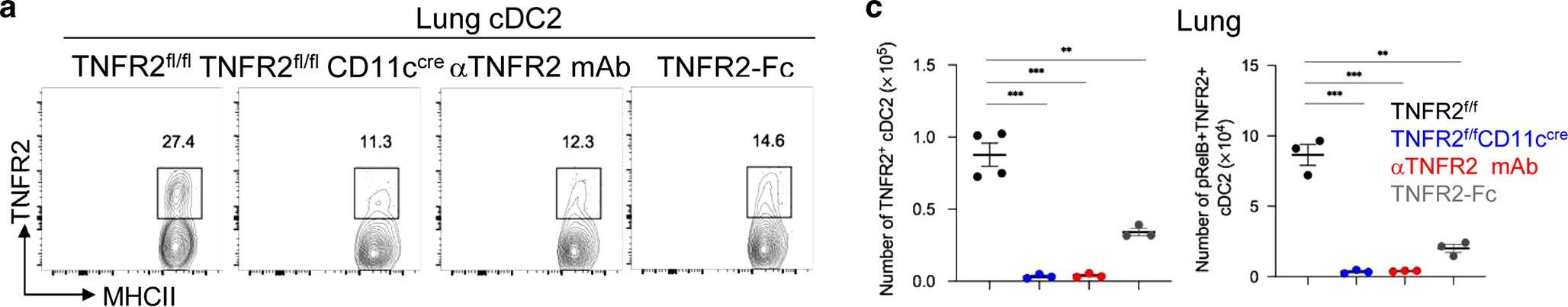
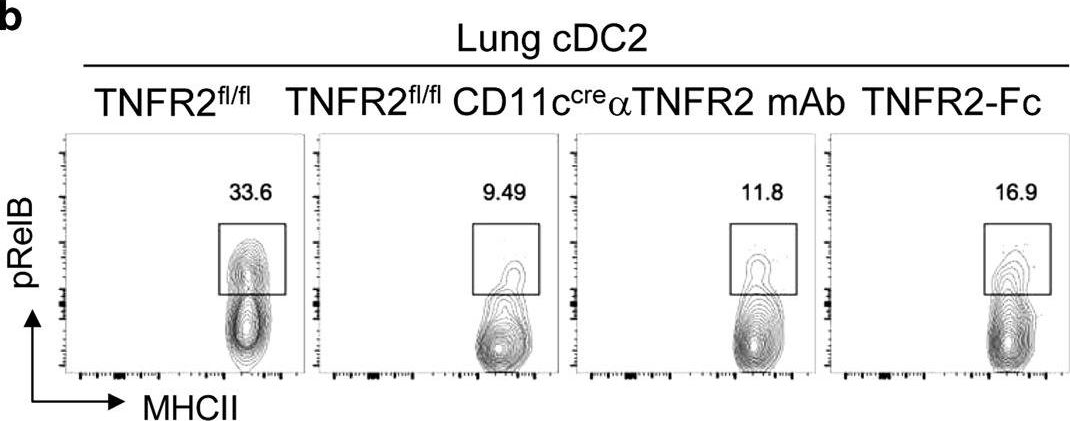
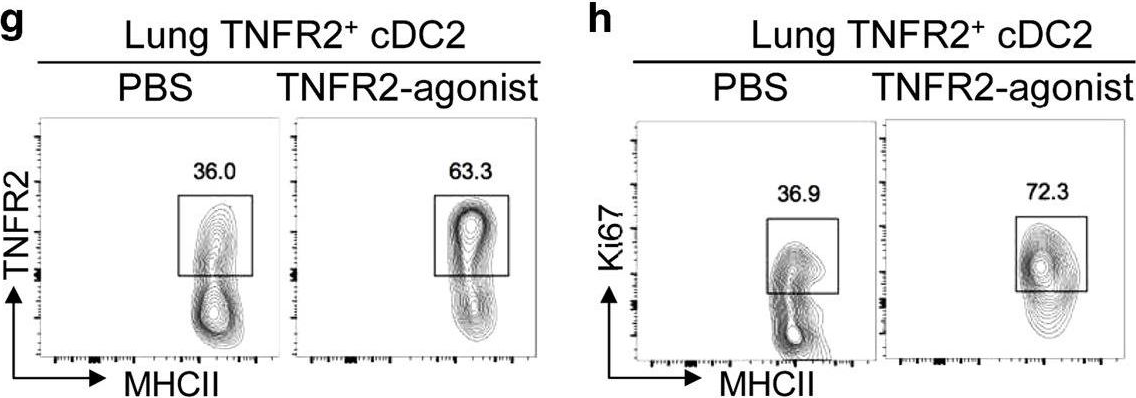
In vivo reprogramming of pathogenic lung TNFR2+ cDC2s by IFNβ inhibits HDM-induced asthma.
In Science Immunology on 9 July 2021 by Mansouri, S., Gogoi, H., et al.
PubMed
Asthma is a common inflammatory lung disease with no known cure. Previously, we uncovered a lung TNFR2+ conventional DC2 subset (cDC2s) that induces regulatory T cells (Tregs) maintaining lung tolerance at steady state but promotes TH2 response during house dust mite (HDM)-induced asthma. Lung IFNβ is essential for TNFR2+ cDC2s-mediated lung tolerance. Here, we showed that exogenous IFNβ reprogrammed TH2-promoting pathogenic TNFR2+ cDC2s back to tolerogenic DCs, alleviating eosinophilic asthma and preventing asthma exacerbation. Mechanistically, inhaled IFNβ, not IFNα, activated ERK2 signaling in pathogenic lung TNFR2+ cDC2s, leading to enhanced fatty acid oxidation (FAO) and lung Treg induction. Last, human IFNβ reprogrammed pathogenic human lung TNFR2+ cDC2s from patients with emphysema ex vivo. Thus, we identified an IFNβ-specific ERK2-FAO pathway that might be harnessed for DC therapy. Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
- Block,
- Mus musculus (House mouse),
- Immunology and Microbiology
The role of neuraminidase in TLR4-MAPK signalling and the release of cytokines by lupus serum-stimulated mesangial cells.
In Immunology on 1 April 2021 by Sundararaj, K., Rodgers, J., et al.
PubMed
Previously, we demonstrated neuraminidase (NEU) activity or NEU1 expression, specifically, is increased in the kidneys of lupus mice and urine of human patients with nephritis. Additionally, NEU activity mediates IL-6 secretion from lupus-prone MRL/lpr primary mouse mesangial cells (MCs) in response to an IgG mimic. IL-6 mediates glomerular inflammation and promotes tissue damage in patients and mouse strains with lupus nephritis. This study further elucidates the mechanisms by which NEU activity and NEU1 specifically mediates the release of IL-6 and other cytokines from lupus-prone MCs. We demonstrate significantly increased release of multiple cytokines and NEU activity in MRL/lpr MCs in response to serum from MRL/lpr mice (lupus serum). Inhibiting NEU activity significantly reduced secretion of three of those cytokines: IL-6, GM-CSF and MIP1α. Message levels of Il-6 and Gm-csf were also increased in response to lupus serum and reduced when NEU activity was inhibited. Neutralizing antibodies to cell-surface receptors and MAPK inhibitors in lupus serum- or LPS-stimulated MCs indicate TLR4 and p38 or ERK MAP kinase signalling play key roles in the NEU-mediated secretion of IL-6. Significantly reduced IL-6 release was observed in C57BL/6 (B6) Neu1+/+ primary MCs compared with wild-type (Neu1+/+) B6 MCs in response to lupus serum. Additional results show inhibiting NEU activity significantly increases sialic acid-containing N-glycan levels. Together, our novel observations support a role for NEU activity, and specifically NEU1, in mediating release of IL-6 from lupus-prone MCs in response to lupus serum through a TLR4-p38/ERK MAPK signalling pathway that likely includes desialylation of glycoproteins. © 2021 John Wiley Sons Ltd.
- Immunology and Microbiology
Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2-expressing Treg cells.
In Proceedings of the National Academy of Sciences of the United States of America on 30 March 2021 by Ronin, E., Pouchy, C., et al.
PubMed
CD4+Foxp3+ regulatory T (Treg) cells are central modulators of autoimmune diseases. However, the timing and location of Treg cell-mediated suppression of tissue-specific autoimmunity remain undefined. Here, we addressed these questions by investigating the role of tumor necrosis factor (TNF) receptor 2 (TNFR2) signaling in Treg cells during experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis. We found that TNFR2-expressing Treg cells were critical to suppress EAE at peak disease in the central nervous system but had no impact on T cell priming in lymphoid tissues at disease onset. Mechanistically, TNFR2 signaling maintained functional Treg cells with sustained expression of CTLA-4 and Blimp-1, allowing active suppression of pathogenic T cells in the inflamed central nervous system. This late effect of Treg cells was further confirmed by treating mice with TNF and TNFR2 agonists and antagonists. Our findings show that endogenous Treg cells specifically suppress an autoimmune disease by acting in the target tissue during overt inflammation. Moreover, they bring a mechanistic insight to some of the adverse effects of anti-TNF therapy in patients.
- In Vivo,
- Mus musculus (House mouse),
- Immunology and Microbiology
Lung IFNAR1hi TNFR2+ cDC2 promotes lung regulatory T cells induction and maintains lung mucosal tolerance at steady state.
In Mucosal Immunology on 1 July 2020 by Mansouri, S., Katikaneni, D. S., et al.
PubMed
The lung is a naturally tolerogenic organ. Lung regulatory T cells (T-regs) control lung mucosal tolerance. Here, we identified a lung IFNAR1hiTNFR2+ conventional DC2 (iR2D2) population that induces T-regs in the lung at steady state. Using conditional knockout mice, adoptive cell transfer, receptor blocking antibodies, and TNFR2 agonist, we showed that iR2D2 is a lung microenvironment-adapted dendritic cell population whose residence depends on the constitutive TNFR2 signaling. IFNβ-IFNAR1 signaling in iR2D2 is necessary and sufficient for T-regs induction in the lung. The Epcam+CD45- epithelial cells are the sole lung IFNβ producer at the steady state. Surprisingly, iR2D2 is plastic. In a house dust mite model of asthma, iR2D2 generates lung TH2 responses. Last, healthy human lungs have a phenotypically similar tolerogenic iR2D2 population, which became pathogenic in lung disease patients. Our findings elucidate lung epithelial cells IFNβ-iR2D2-T-regs axis in controlling lung mucosal tolerance and provide new strategies for therapeutic interventions.
- In Vivo,
- Mus musculus (House mouse)
TNFR2 Signaling Enhances ILC2 Survival, Function, and Induction of Airway Hyperreactivity.
In Cell Reports on 24 December 2019 by Hurrell, B. P., Galle-Treger, L., et al.
PubMed
Group 2 innate lymphoid cells (ILC2s) can initiate pathologic inflammation in allergic asthma by secreting copious amounts of type 2 cytokines, promoting lung eosinophilia and airway hyperreactivity (AHR), a cardinal feature of asthma. We discovered that the TNF/TNFR2 axis is a central immune checkpoint in murine and human ILC2s. ILC2s selectively express TNFR2, and blocking the TNF/TNFR2 axis inhibits survival and cytokine production and reduces ILC2-dependent AHR. The mechanism of action of TNFR2 in ILC2s is through the non-canonical NF-κB pathway as an NF-κB-inducing kinase (NIK) inhibitor blocks the costimulatory effect of TNF-α. Similarly, human ILC2s selectively express TNFR2, and using hILC2s, we show that TNFR2 engagement promotes AHR through a NIK-dependent pathway in alymphoid murine recipients. These findings highlight the role of the TNF/TNFR2 axis in pulmonary ILC2s, suggesting that targeting TNFR2 or relevant signaling is a different strategy for treating patients with ILC2-dependent asthma. Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
- In Vivo,
- Mus musculus (House mouse),
- Biochemistry and Molecular biology,
- Immunology and Microbiology
A temporally dynamic Foxp3 autoregulatory transcriptional circuit controls the effector Treg programme.
In The EMBO Journal on 15 August 2018 by Bending, D., Paduraru, A., et al.
PubMed
Regulatory T cells (Treg) are negative regulators of the immune response; however, it is poorly understood whether and how Foxp3 transcription is induced and regulated in the periphery during T-cell responses. Using Foxp3-Timer of cell kinetics and activity (Tocky) mice, which report real-time Foxp3 expression, we show that the flux of new Foxp3 expressors and the rate of Foxp3 transcription are increased during inflammation. These persistent dynamics of Foxp3 transcription determine the effector Treg programme and are dependent on a Foxp3 autoregulatory transcriptional circuit. Persistent Foxp3 transcriptional activity controls the expression of coinhibitory molecules, including CTLA-4 and effector Treg signature genes. Using RNA-seq, we identify two groups of surface proteins based on their relationship to the temporal dynamics of Foxp3 transcription, and we show proof of principle for the manipulation of Foxp3 dynamics by immunotherapy: new Foxp3 flux is promoted by anti-TNFRII antibody, and high-frequency Foxp3 expressors are targeted by anti-OX40 antibody. Collectively, our study dissects time-dependent mechanisms behind Foxp3-driven T-cell regulation and establishes the Foxp3-Tocky system as a tool to investigate the mechanisms behind T-cell immunotherapies. © 2018 The Authors. Published under the terms of the CC BY 4.0 license.
- Biochemistry and Molecular biology,
- Immunology and Microbiology
A temporally dynamic i>Foxp3/i> autoregulatory transcriptional circuit controls the effector Treg programme
Preprint on BioRxiv : the Preprint Server for Biology on 10 January 2018 by Bending, D., Paduraru, A., et al.
PubMed
Regulatory T cells (Treg) are negative regulators of the immune response. Whilst thymic Treg generation is well studied, it is not known whether and how Foxp3 transcription is induced and regulated in the periphery during immune responses. Here we use Foxp3 T imer o f c ell k inetics and activit y (Tocky) mice, which report real-time Foxp3 gene transcription by measuring the spontaneous maturation of Fluorescent Timer protein from Blue to Red fluorescence, to identify the flux of Foxp3 -to Foxp3 + T cells within the periphery and analyse the real-time activity of Foxp3 transcription. Using a murine model of skin allergy, we show that both the flux of new Foxp3 expressors and the rate of Foxp3 transcription are increased at inflamed sites. These persistent dynamics of Foxp3 transcription determine the effector Treg programme, and are dependent on a Foxp3 autoregulatory transcriptional circuit, as evidenced by analysis of T cells lacking functional Foxp3 protein. Such reactive and persistent Foxp3 transcriptional activity controls the expression of coinhibitory molecules including CTLA-4 and effector-Treg signature genes. Using RNA-seq, we identify two groups of surface proteins based on their relationship to the temporal dynamics of Foxp3 transcription, and we show proof-of-principle for the manipulation of Foxp3 dynamics by immunotherapy: new Foxp3 flux is promoted by anti-TNFRII antibody, and high frequency Foxp3 expressors are depleted by anti-OX40 antibody. Collectively, our study dissects time-dependent mechanisms behind Foxp3-driven T cell regulation, and establishes the Foxp3-Tocky system as a tool to investigate the mechanisms behind T cell immunotherapies.
- Cancer Research,
- Immunology and Microbiology
TNFR2/BIRC3-TRAF1 signaling pathway as a novel NK cell immune checkpoint in cancer.
In Oncoimmunology on 11 October 2017 by Ivagnes, A., Messaoudene, M., et al.
PubMed
Natural Killer (NK) cells control metastatic dissemination of murine tumors and are an important prognostic factor in several human malignancies. However, tumor cells hijack many of the NK cell functional features compromising their tumoricidal activity. Here, we show a deleterious role of the TNFα/TNFR2/BIRC3/TRAF1 signaling cascade in NK cells from the tumor microenvironment (TME). TNFα induces BIRC3/cIAP2 transcripts and reduces NKp46/NCR1 transcription and surface expression on NK cells, promoting metastases dissemination in mice and poor prognosis in GIST patients. NKp30 engagement, by promoting the release of TNFα, also contributes to BIRC3 upregulation, and more so in patients expressing predominantly NKp30C isoforms. These findings reveal that in the absence of IL-12 or a Th1-geared TME, TNFα can be considered as a negative regulatory cytokine for innate effectors.
- In Vivo,
- Mus musculus (House mouse),
- Cardiovascular biology,
- Immunology and Microbiology
Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells.
In Blood on 22 September 2016 by Leclerc, M., Naserian, S., et al.
PubMed
Therapeutic CD4(+)Foxp3(+) natural regulatory T cells (Tregs) can control experimental graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation (allo-HCT) by suppressing conventional T cells (Tconvs). Treg-based therapies are currently tested in clinical trials with promising preliminary results in allo-HCT. Here, we hypothesized that as Tregs are capable of modulating Tconv response, it is likely that the inflammatory environment and particularly donor T cells are also capable of influencing Treg function. Indeed, previous findings in autoimmune diabetes revealed a feedback mechanism that renders Tconvs able to stimulate Tregs by a mechanism that was partially dependent on tumor necrosis factor (TNF). We tested this phenomenon during alloimmune response in our previously described model of GVHD protection using antigen specific Tregs. Using different experimental approaches, we observed that control of GVHD by Tregs was fully abolished by blocking TNF receptor type 2 (TNFR2) or by using TNF-deficient donor T cells or TNFR2-deficient Tregs. Thus, our results show that Tconvs exert a powerful modulatory activity on therapeutic Tregs and clearly demonstrate that the sole defect of TNF production by donor T cells was sufficient to completely abolish the Treg suppressive effect in GVHD. Importantly, our findings expand the understanding of one of the central components of Treg action, the inflammatory context, and support that targeting TNF/TNFR2 interaction represents an opportunity to efficiently modulate alloreactivity in allo-HCT to either exacerbate it for a powerful antileukemic effect or reduce it to control GVHD. © 2016 by The American Society of Hematology.